

论文拟解决的关键挑战:
当前,阴离子交换膜燃料电池(AEMFCs)中非贵金属(PGM-free)阳极催化剂的发展面临严峻挑战:虽然镍(Ni)是目前唯一展现出氢氧化反应(HOR)活性的非铂族金属,但其本征活性有限,电化学活性面积(ECSA)显著低于贵金属催化剂,导致整体催化性能不足。现有最佳Ni基阳极的峰值功率密度(PPD)普遍低于630 mW cm⁻²,远未达到实用化要求。更关键的是,传统改性策略如合金化、表面修饰和金属-载体相互作用(MSI)仅能激活催化剂表面极少量的Ni原子,大部分Ni原子仍处于惰性状态,活性位点密度严重不足。如何在不牺牲本征活性的前提下,大幅提升活性位点密度,并系统阐明多层原子级电子调控机制,成为推动PGM-free AEMFCs发展的核心难题。
针对上述挑战,中国科学技术大学的陈乾旺教授和以色列理工学院的Dario R. Dekel教授等人在Energy & Environmental Science发表论文,该团队创新性地提出了多层近邻原子激活策略,通过构建Fe单原子掺杂的Ni基合金催化剂(Fe₁NiSAA-O@CN),成功将Fe周围第一至第三近邻的Ni原子均激活为高活性位点,在保持高本征活性的同时显著提升了活性位点密度,实现了AEMFC性能的突破性进展。
催化剂精准设计与可控合成
作者采用创新的L-谷氨酸配位前驱体路线,通过精确调控Fe²⁺和Ni²⁺的摩尔比例,在400°C的H₂/Ar还原气氛下进行热解,成功制备出具有明确单原子分散特征的Fe₁Ni合金催化剂。系统合成了不同Fe含量的对比样品(包括Ni-O@CN、Fe₅Ni₉₅-O@CN、FeNi₃-O@CN等),建立了成分-结构-性能的构效关系,确定了最优Fe掺杂量为6.85 wt%。合成过程中形成的多孔氮掺杂碳载体不仅作为高效的电子传导网络,还起到了纳米反应器的作用,有效防止了活性组分的溶解和团聚。
多层次结构表征与电子结构解析
晶体结构分析:XRD显示Fe掺杂导致Ni(111)晶面衍射角负移,晶面间距从0.203 nm扩大至0.209 nm,直接证明了Fe原子成功进入Ni晶格。
微观形貌表征:HRTEM显示合金纳米颗粒尺寸均匀分布在~7.4 nm,且清晰观察到Ni(111)和C(002)晶格条纹,证实了催化剂的高度结晶性。
元素分布与化学状态:HAADF-STEM结合EDS mapping显示了Fe、Ni、C、N元素的均匀分布,线扫描分析进一步证实了Fe在Ni基质中的原子级分散。
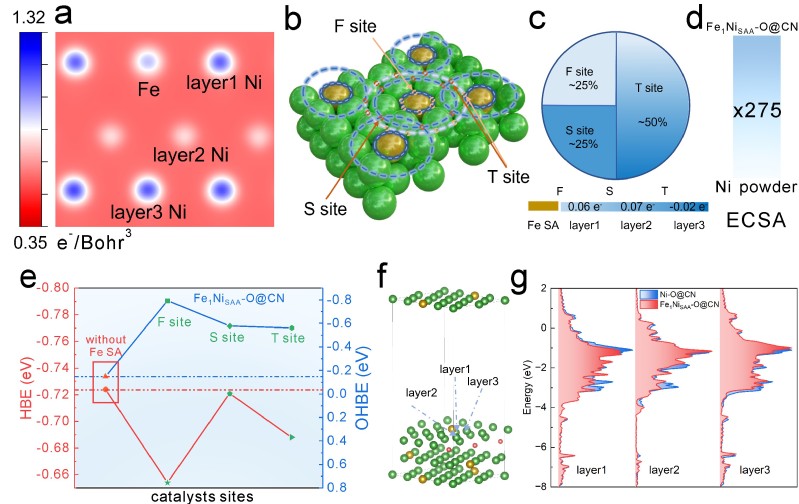
原子级电子结构研究:创新性地采用原子分辨率EELS技术,通过对Fe L₃边和Ni L₃边的线扫描分析,首次在实验上观察到Fe单原子对其周围三层Ni原子的电子调控作用,Ni L₃边结合能从856.0 eV负移至854.5 eV,证明电子从Fe向Ni转移。
表面电子性质与配位环境深度分析
XPS分析显示Ni 2p轨道结合能负移,而Fe 2p轨道正移,直接证实了电子从Fe向Ni转移的方向,这一结果与UPS测得的功函数差异完全吻合。同步辐射XAFS研究提供了更深入的配位环境信息:Ni K边XANES表明Ni保持金属态,而Fe K边XANES显示Fe的氧化态与酞菁铁相似,证明其单原子分散特征。EXAFS拟合和Wavelet变换分析进一步确认了Fe-Ni配位键的存在,排除了Fe-Fe配位的可能性,为单原子合金结构提供了确凿证据。
电化学性能系统评估与机理探究
碱性HOR性能:在0.1 M KOH中,Fe₁NiSAA-O@CN的质量活性高达67.58AgNi⁻¹,交换电流密度(j₀)达1.85 mA cm⁻²,远超商业Pt/C和其他对比样品。
活性位点密度提升:ECSA达到37.1m²gNi⁻¹,是传统Ni粉末的275倍,创造了Ni基HOR催化剂的最高纪录。
稳定性与耐受性:经过5000次循环加速测试后仍保持72.4%的初始活性,显著优于Pt/C的17.2%。同时展现出优异的CO耐受性,在1000 ppm CO存在下性能衰减小于10%。
反应机理研究:通过微极化区拟合和Butler-Volmer方程计算,系统分析了反应动力学参数,证实了Fe掺杂显著提升了本征反应速率。
AEMFC实际运行性能验证
单电池性能突破:使用Fe₁NiSAA-O@CN作为阳极的H₂/O₂燃料电池,峰值功率密度达到729.4 mW cm⁻²,是目前文献报道的Ni基阳极最高值,比商业Ni粉提升4.4倍。
全非贵金属电池实现:与PGM-free阴极催化剂CoFe/CDC/CNT配对,在H₂/O₂条件下PPD达420.9 mW cm⁻²,在H₂/无CO₂空气条件下达271.4 mW cm⁻²,展现了全PGM-free AEMFC的商业化潜力。
长期稳定性测试:在200 mA cm⁻²恒流放电条件下连续运行100小时,电压衰减率仅为1.26 mV h⁻¹,展现了优异的实际应用前景。
理论计算与机制深入阐释
电子结构计算:DFT计算显示Fe向其周围三层Ni原子转移电子,Bader电荷分析定量给出了电子转移数:第一近邻0.06 e⁻,第二近邻0.07 e⁻,第三近邻0.02 e⁻。
吸附能调控机制:系统计算了不同近邻位点的H和OH吸附能,发现Fe掺杂使所有近邻Ni位点的HBE减弱而OHBE增强,实现了吸附能的双向优化。
态密度分析:通过投影态密度(PDOS)计算,揭示了Fe掺杂引起Ni的d带中心上移,从理论上解释了吸附能变化的电子结构根源。
活性位点统计:理论模型表明,在理想情况下,第一、二、三近邻Ni原子分别占25%、25%和50%,意味着大部分表面Ni原子都被激活,完美解释了ECSA的大幅提升。
本研究首次提出并实验验证了三层近邻原子激活概念,突破了传统催化设计中仅考虑最近邻原子的局限。
建立了从原子级结构设计到宏观电池性能的完整研究范式,为新型PGM-free催化剂开发提供了全新思路。
论文链接:Energy Environ. Sci., DOI: 10.1039/d5ee04644k